
What America Needs from a New FDA Commissioner
Beat China, Unleash Cures.

Dear Readers,
At 8VC, we take a strong interest in the Food and Drug Administration as entrepreneurs and builders who’ve been deeply involved in the US biotech sector for decades. Last year during the first 100 days of the new Trump administration, we published a lot of ideas for the agency.
You may have read earlier this month that Dr. Marty Makary departed as FDA Commissioner. Now, as the administration seeks a new leader for the FDA, we wanted to share with you some of the principles and frameworks that we are circulating in Washington.
Every administration has to make tough decisions amid a lot of private jockeying and feedback from various interests (not all of them honest or positively-aligned), so it’s a little unorthodox to do this in public. But after all, the FDA isn’t “for” Washington — it’s for you! More than 300 million Americans need a bold, courageous FDA because we can do so much better as a country in bringing new cures to our fellow citizens who need them most.
So, in that spirit we’re publishing the issues that we think are key right now.
One of the most gratifying parts of our work as investors is seeing the brightness of the future come into focus. In just the last month we’ve seen data from an amazing new gene therapy lowering cholesterol heart attack risk, and another attacking pancreatic cancer. Whether these breakthroughs reach patients in time will depend not only on the scientists and founders building them, but on whether the FDA has a functional, innovative culture.
As ever, it is strong leadership that ultimately matters most.
Joe
More than any other in recent memory, this administration has been willing to be bold in defense of American interests. It must do so for US biotech as well. Our US biotech ecosystem is in crisis: to be blunt, China’s industrial policy combined with a sclerotic FDA over the previous decade is stealing and eroding away the industry. This is happening at a time when AI breakthroughs should be accelerating faster and cheaper cures.
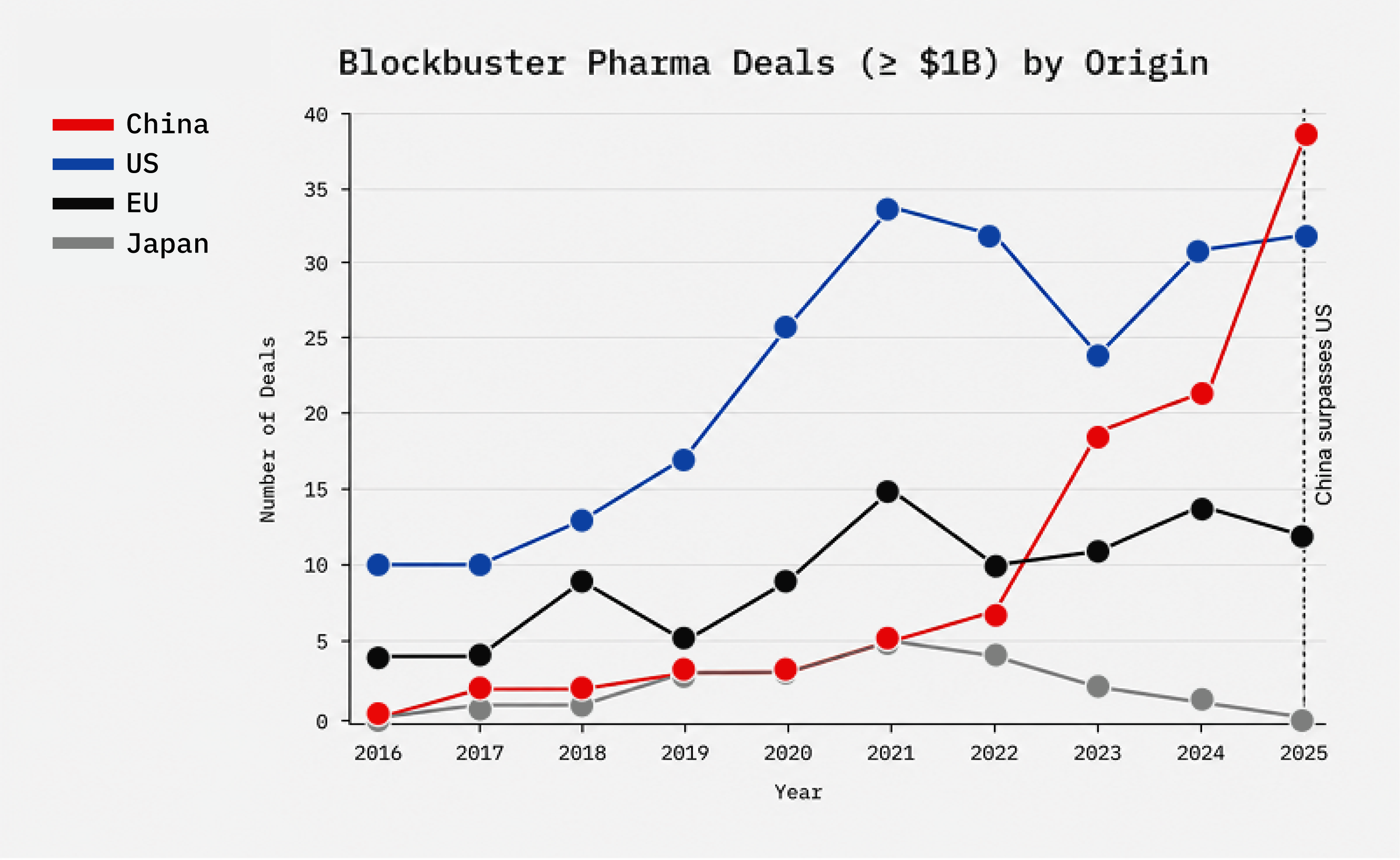
China is not “competing” with American biotech, this isn’t a level playing field. The Chinese Communist Party (CCP) is flooding its companies with capital, tilting rules for domestic champions, and leaning on IP theft and coercion.
To fix this, the next leader of the FDA must be a fighter. This includes not just embracing innovation but also confronting and at times overruling the internal bureaucracy. The next commissioner can do all of this by prioritizing speed, domestic resiliency, and regulatory clarity across drugs and clinical AI.
As the administration seeks a new commissioner, we think these topics make for great starting points in their interviews about their plans.
1. Accelerate generation of US clinical data on a level playing field
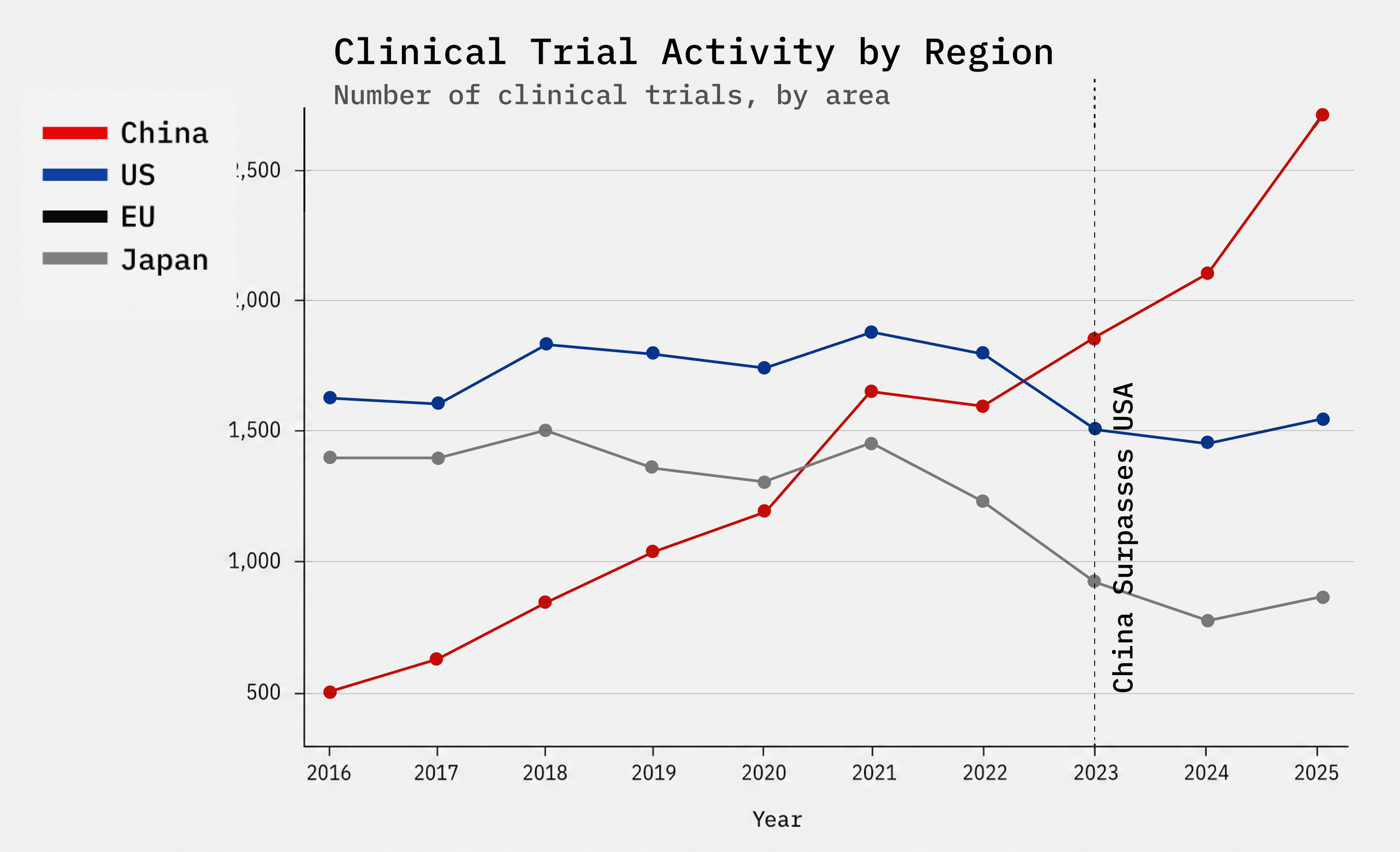
A. Create an expedited Investigational New Drug (IND) pathway for Phase 1 trials. Our current Phase 1 trial regulations require 12–18 months of preclinical work that Australia’s version of an IND (in Australia called a CTN) have shown unnecessary; the US regulations are a top-down, Soviet legacy of the mid-20th century that is straitjacketing us and delaying innovation. If we implement the Australian learnings, including 3rd party or medical center trial oversight (who review the IND instead of the FDA) we can speed up patient access to novel drugs while saving FDA reviewers’ time and taxpayer dollars.
The leading research institutions of the world like MD Anderson Cancer Center and Memorial Sloan Kettering should be empowered to decide when and how they launch Phase 1 trials, within regulatory frameworks.
B. Scale and operationalize FDA’s Real-time clinical trials initiative to reduce the dead time in-between clinical phases. Current clinical workflows are discontinuous with weeks to months of dead time between phases. Scale the pilot into a broad initiative to monitor trials and trigger reviewer actions or feedback on trial designs quickly.
C. Ensure foreign clinical, preclinical and manufacturing sites are at US standards. Continue prioritizing foreign inspections and enforce in-person inspections with no warning. Ex-US sites, in particular those in China, have serious ethical issues (such as consent) in China Investigator Initiated Trials or trials in Xinjiang province. Chinese animal testing companies engage in work which would be illegal under US animal welfare laws. Ensure that US researchers aren’t penalized for following US rules through robust ex-US enforcement to produce a level playing field at all scientific stages.
D. Codify and harden objective criteria for the Commissioner’s National Priority Voucher that require US based preclinical work, manufacturing & primary US clinical sites as a condition for receiving a voucher.
2. Create modern frameworks for AI regulations
A. Create a predictable pathway for clinical AI approval and monitoring. Clinical AI systems need clear benchmarks aligned to outcomes that matter to patients. FDA should prescriptively develop reasonable approval bars with post approval monitoring and an eye towards keeping innovation speed equal to safety concerns alongside a culture that partners with US innovators.
B. Enable controlled continuous learning. AI systems get better through continuous learning. Legacy regulations focused on single point in time evaluations and should instead enable the continuous nature of these systems so that patients can benefit from the latest and greatest data; not those from 12 months ago.
3. Better align approval and commercial requirements to patient needs
A. Create tiered commercial Good Manufacturing Practice (GMP) standards matched to patient need. FDA rules today apply different clinical standards for different patient populations based on need. But GMP standards are the same for all. Define fit-for-purpose requirements for n-of-1, ultra-rare and broad-population products, preserving safety-critical controls while avoiding large commercial-scale burdens where they do not substantially impact patient safety for small product lots.
B. Approve more surrogate endpoints, or other alternative clinical data sources Whenever the FDA formally approves a new short-term surrogate endpoint for drug development massive capital and innovation flow to find cures for these diseases. The progression-free-survival endpoint drove tons of advances in cancer that we are reaping today (from PD-1 antibodies to curative cell therapies). FDA needs to lean into this superpower and accelerate qualification of more surrogate endpoints and beyond into more acceptance of natural-history datasets and digital endpoints that can better enable rural patient outcomes in the approval decision making frameworks. FDA must also finish the job on the Plausible Mechanism Framework for individualized therapies.
C. Convince companies they won’t be penalized for providing right-to-try or expanded access. Today many pharmas and biotechs provide little to no opportunities to access early clinical phase novel therapies under the existing right-to-try and expanded access rules. This happens since companies perceive a strong disincentive by the FDA: adverse events from either pathway must be reported to the FDA (and thus counts against your drug) but they fear the FDA ignoring positive signals from those very same patients that prove the drug works. The FDA must remove this fear from companies by providing ironclad clarity that the positive data will count.
The US can and should be the fastest, safest place in the world to generate clinical evidence, deploy clinical AI, and get promising cures to patients. If these recommendations and more from other smart experts are implemented, the future would be very bright indeed. Billions of dollars of capital would flow to US biotechs, driving a scientific renaissance that in turn saves and improves many lives.

Yes Sir, thank you for all you’re doing Joe.
There’s much to agree with here especially the adoption of an Aussie style CTN and the expansion of the real-time reporting approach currently in pilot mode.
Enhanced biomarker utilisation also sounds good but genuine biomarker validation is not trivial.
Taken as a whole this suggests that the next head of the FDA needs to be someone comfortable with more unorthodox thinking (but that does not mean a lack of rigour).